High Performance Computing
In the era of big data, we need big computers. Just like other expensive resources these are shared among many researchers, however this also makes them easy to get access to. At UT Dallas alone we have a few clusters you can utilize:
- Sysbio, for training
- Ganymede, for High Performance Computing
- Europa(Public access coming soon), for High Throughput Computing
Just for comparison, a node in the genomics queue on Ganymede has 16 cores, 32GB of RAM, and is connected to a petabyte of storage. There's also 16 nodes. They're free for you to use as well! You just need to know how to use them.
What's a cluster?
- You login to the login node
- Login nodes are a prep area
- Transferring files
- Compiling code
- Submitting jobs
- The compute nodes are where actual computations occur and where research is done. All batch jobs and executables, as well as development and debugging sessions, are run on the compute nodes.1
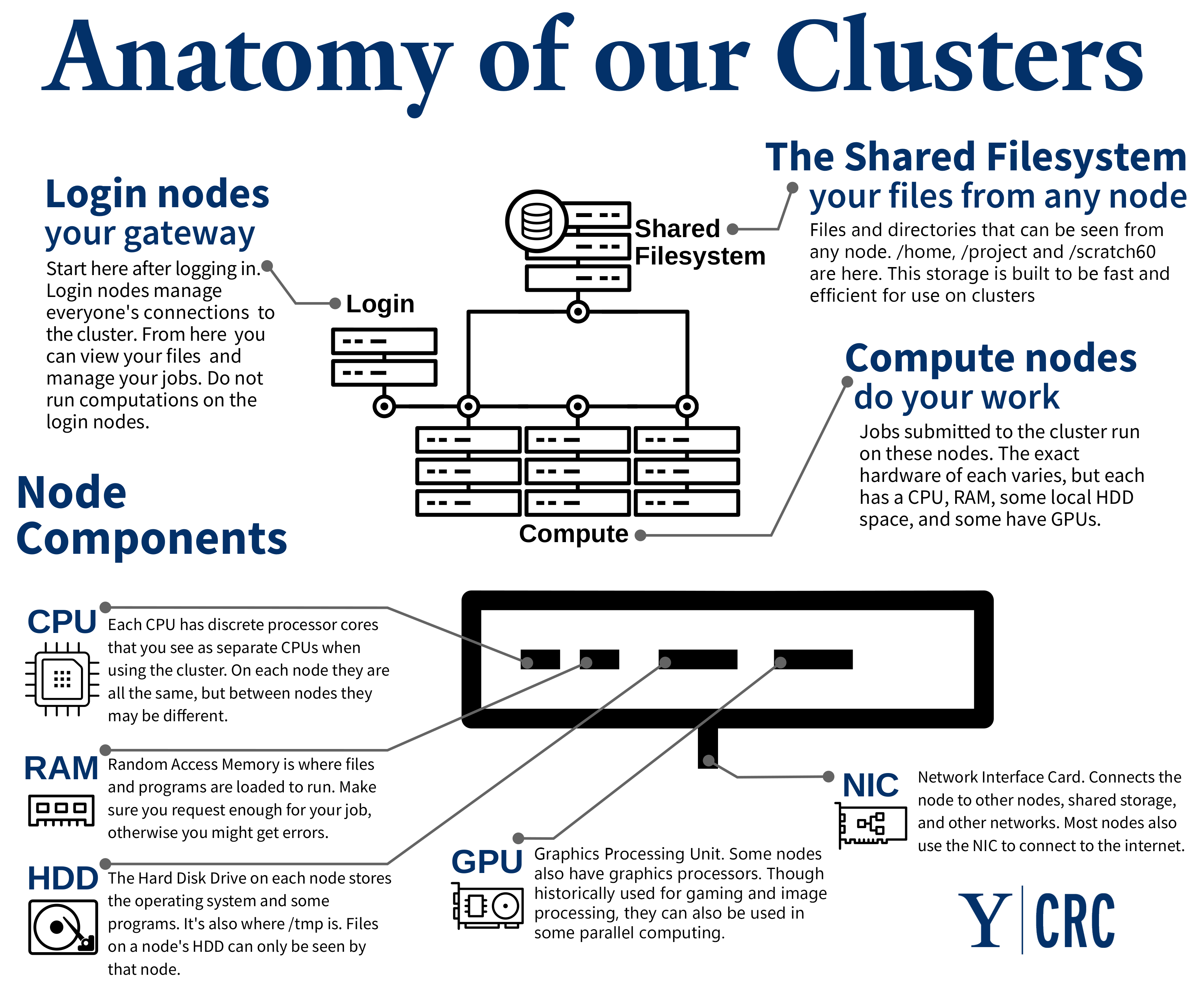
- The nodes all have access to a shared filesystem
Slurm
SLURM - Simple Linux Utility for Resource Management
Provides a way for users to schedule jobs. There are plenty of alternatives but Slurm is pretty common. Checkout this Slurm Cheatsheet if you need to reference the commands to control it.
Tutorial
info
Before we start with our first steps tutorial, we would like to introduce the following convention that we use throughout the series:
$ Commands are prefixed with a little dollar sign
login $ The host name is put infront of the dollar sign
compute $
Instant Gratification
login $ srun -p normal --time 1-00 --mem=8G --ntasks=8 --pty bash -i
# Once you're logged into a node
compute $ exit
scratch
All users on the system have a file system quota. You'll see it every time you
login. There's a soft limit, that if you go over you'll get the warning: dm-0: warning, user block quota exceeded.. There's also a Hard limit, which if
you exceed it the process will be killed. You can check your quota with quota -s
See Ganymede Space Constraints for more info
But you should notice in your home directory you have a symbolic link called
scratch. This is connected to a large storage system and is unmetered.
Here's a quick table comparing the two:
| Attributes | Home Space | Scratch Space |
|---|---|---|
| Purpose of the Space | To house scripts, source code and output data. | To have a large area for working with data or trying ideas |
| Backed Up | Yes | No |
Let's get some data
So how do we get data from our browser, to the computer? How do we avoid downloading it locally(to our laptops, which have limited space)?
wget-The non-interactive network downloader
- wget allows us to download files from the web via the command line
- It supports http, https and ftp
Let's use it!
- Let's clone our project into the scratch directory
login $ cd scratch
login $ git clone https://github.com/github-username/ag-intro.git
login $ cd ag-intro
- Download the data.
login $ wget https://github.com/nf-core/test-datasets/raw/modules/data/genomics/homo_sapiens/genome/genome.gtf
login $ wget https://github.com/nf-core/test-datasets/raw/modules/data/genomics/homo_sapiens/illumina/bam/test.paired_end.sorted.bam
While we're at it, let's through these commands into our Makefile so we don't forget the commands!
download_data: genome.gtf test.paired_end.sorted.bam
genome.gtf:
wget https://github.com/nf-core/test-datasets/raw/modules/data/genomics/homo_sapiens/genome/genome.gtf
test.paired_end.sorted.bam:
wget https://github.com/nf-core/test-datasets/raw/modules/data/genomics/homo_sapiens/illumina/bam/test.paired_end.sorted.bam
Don't forget to add the data to .gitignore before you commit!
What did we just download?
These files are two different formats that are common:
The GFF/GTF/BED formats are the so-called “interval” formats that retain only the coordinate positions for a region in a genome. Each is tab delimited and will contain information on the chromosomal coordinate, start, end, strand, value and other attributes, though the order of the columns will depend on the feature.4
The GFF/GTF formats are 9 column tab-delimited formats. The coordinates start at value 1.
chr1 . mRNA 1300 9000 . + . ID=mrna0001;Name=sonichedgehog
chr1 . exon 1300 1500 . + . ID=exon00001;Parent=mrna0001
chr1 . exon 1050 1500 . + . ID=exon00002;Parent=mrna0001
The SAM/BAM formats are so-called Sequence Alignment Maps. These files typically represent the results of aligning a FASTQ file to a reference FASTA file and describe the individual, pairwise alignments that were found. Different algorithms may create different alignments (and hence BAM files).4
SRR1553425.13617 163 AF086833.2 46 60 101M = 541 596 GAATAACTATGAGGAAGATTAATAATTTTCCTCTCATTGAAATTTATATCGGAATTTAAATTGAAATTGTTACTGTAATCATACCTGGTTTGTTTCAGAGC @<@FFFDDHHGHHJIJJIJJJJJJJJJJJJJJIIJJJJGIJJJJJJEIEIGIIJJJJJIIIIGIIIGGEGIIIGIIJJJGGGHHFHGFDDFDEEDDEDCDCNM:i:1 MD:Z:81C19 MC:Z:101M AS:i:96 XS:i:0
SRR1553425.13755 99 AF086833.2 46 60 101M = 46 101 GAATAACTATGAGGAAGATTAATAATTTTCCTCTCATTGAAATTTATATCGGAATTTAAATTGAAATTGTTACTGTAATCATACCTGGTTTGTTTCAGAGC ?:@DBDDDFHGDHGIGEGAFGGHDHIIIIIIIGGGGIGEBHHGHFGHIDCHIIIIII<G@FGHDFGGHH@FGGEGGGEHIE@HHHGHF;?BC@CECFC:@>NM:i:1 MD:Z:81C19 MC:Z:101M AS:i:96 XS:i:0
SRR1553425.13755 147 AF086833.2 46 60 101M = 46 -101 GAATAACTATGAGGAAGATTAATAATTTTCCTCTCATTGAAATTTATATCGGAATTTAAATTGAAATTGTTACTGTAATCATACCTGGTTTGTTTCAGAGC DDDDCDDDDDECEEDEDFFFEFFGHHHHIGGHG@=@@=GIIJHIIHFDIIIIIIHHGJIFAJJJIGIJIIIJHHGGIGEGGHFHHJIIHHHGHFFFFFCCCNM:i:1 MD:Z:81C19 MC:Z:101M AS:i:96 XS:i:0
SRR1553425.11219 2227 AF086833.2 47 60 71H30M = 146 171 AATAACTATGAGGAAGATTAATAATTTTCC IGHF@GHEHIIHE>IIGHFDHDDDFFF@@@ NM:i:0 MD:Z:30 MC:Z:101M AS:i:30 XS:i:0 SA:Z:AF086833.2,68,+,30S71M,60,1;
You can find more info at Biostars handbook Common data types
Some other ways to get data onto the cluster
wget isn't the only way to get things to the cluster. It's great for stuff that's on the internet, but what about local files?5
SCP - secure copy
- Local file to remote server
scp filename.txt hostname:/path/to/filename/filename.txt
- Remote server to local file
scp hostname:filename.txt .
- Recursive copies on directories also work with
-r
scp –r hbar.ices.utexas.edu:public_html ~/tmp
rsync - a fast, versatile, remote (and local) file-copying tool
- Example 1. Synchronize Two Directories in a Local Server
rsync -zvr /path/to/source /path/to/copy
z- enable compressionv- verboser-recursiveExample 2. Preserve timestamps during Sync using rsync
–a
rsync -azv /path/to/source /path/to/copy
-aenters archive mode- Archive mode does the following:
- Recursive mode
- Preserves symbolic links
- Preserves permissions
- Preserves timestamp
- Preserves owner and group
Back to the Tutorial
So we've got the data. But how do we get the software we need to do anything with it?
We'll first use Lmod: A New Environment Module System
Some helpful commands:
login $ module list # What modules do I currently have enabled?
login $ module load subread # Load the subread module
login $ module unload subread # Unload the subread module
login $ module unload anaconda3 # Load the anaconda3 module
login $ module save default # Save the current module stack to the default stack
So let's actually run featurecounts and get the counts for our genes!
login $ srun -p normal --time 1-00 --mem=8G --ntasks=8 --pty bash -i
compute $ ml load subread
compute $ featureCounts -t exon -g gene_id -a genome.gtf -o counts.txt test.paired_end.sorted.bam
Pop open counts.txt with cat counts.txt and check out the data!
sbatch
So the interactive login for jobs is great, when no one else is using the cluster! Why should we have to wait around in line all day to get our time on a compute node? What if we could tell the cluster what we wanted to do and it would schedule the jobs for us?
You use the sbatch command in front of the script you actually want to run. sbatch then puts your job into the job queue. The job scheduler looks at the current status of the whole system and will assign the first job in the queue to a node that is free in terms of computational load. If all machines are busy, yours will wait. But your job will sooner or later get assigned to a free node. 6
Example script
#!/bin/bash
# Set a name for the job (-J or --job-name).
#SBATCH --job-name=tutorial
# Set the file to write the stdout and stderr to (if -e is not set; -o or --output).
#SBATCH --output=logs/%x-%j.log
# Set the number of cores (-n or --ntasks).
#SBATCH --ntasks=8
# Force allocation of the two cores on ONE node.
#SBATCH --nodes=1
# Set the total memory. Units can be given in T|G|M|K.
#SBATCH --mem=8G
# Optionally, set the partition to be used (-p or --partition).
#SBATCH --partition=medium
# Set the expected running time of your job (-t or --time).
# Formats are MM:SS, HH:MM:SS, Days-HH, Days-HH:MM, Days-HH:MM:SS
#SBATCH --time=30:00
export TMPDIR=/fast/users/${USER}/scratch/tmp
mkdir -p ${TMPDIR}
info
The lines starting with #SBATCH are actually setting parameters for a sbatch command, so #SBATCH --job-name=tutorial is equal to sbatch --job-name=tutorial.
Turning our featurecount command into a script
Open up a new file in your text editor
#!/bin/bash
#SBATCH --job-name=featurecounts
#SBATCH --ntasks=8
#SBATCH --nodes=1
#SBATCH --mem=8G
#SBATCH --partition=normal
#SBATCH --time=30:00
ml load subread
featureCounts -t exon -g gene_id -a genome.gtf -o counts.txt test.paired_end.sorted.bam
And then submit it to the cluster with
login $ sbatch submit_job.sh
login $ cat *.out
Commit your Changes!
Make sure to gitignore your input data and any results you produced! Along with
any log files from sbatch.
It's up to you whether you want to create a make rule for sbatch or just include it in the README
Further Reading
- TACC Lonestar5 User guide↩
- Yale HPC Docs↩
- UTDallas HPC Cluster Users Guide↩
- Biostars handbook Common data types↩
- [HPC Class SSH and Slurm lecture]↩
- BIH HPC Docs↩